Models (Introduction)
This is an introduction to models in TeNPy, intended to guide new-comers towards defining their own custom models.
We go step by step to introduce the relevant concepts and “derive” how you could have come up with the following example code to implement an anisotropic Heisenberg model on a square lattice:
class MyModel(CouplingMPOModel):
r"""The anisotropic spin-1/2 Heisenberg model in an external field.
This is a pedagogical example, and you should probably use the SpinModel instead.
The Hamiltonian is:
.. math ::
H = J_x \sum_i (S^x_i S^x_{i+1} + S^y_i S^y_{i+1})
+ J_z \sum_i S^z_i S^z_{i+1}
- h_x \sum_i S^z_i - h_z \sum_i S^z_i
"""
def init_sites(self, model_params):
conserve = model_params.get('conserve', 'best')
if conserve == 'best':
if model_params.get('hx', 0) == 0:
conserve = 'Sz'
else:
conserve = None
return SpinHalfSite(conserve=conserve)
def init_terms(self, model_params):
Jx = model_params.get('Jx', 1.)
Jz = model_params.get('Jz', 1.)
hx = model_params.get('hx', 0.)
hz = model_params.get('hz', 0.)
for u in range(len(self.lat.unit_cell)):
self.add_onsite(-hx, u, 'Sx')
self.add_onsite(-hz, u, 'Sz')
for u1, u2, dx in self.lat.pairs['nearest_neighbors']:
self.add_coupling(Jz, u1, 'Sz', u2, 'Sz', dx)
self.add_coupling(.5 * Jx, u1, 'Sp', u2, 'Sm', dx, plus_hc=True)
model = MyModel({'lattice': 'Square', 'Lx': 2, 'Ly': 4, 'Jx': 0})
What is a model?
Abstractly, a model stands for some physical (quantum) system, described by a Hamiltonian. For example, let us consider an anisotropic spin-1/2 Heisenberg model in a field, described by
The main features that need to be defined for a model are
The local Hilbert space. In this example it is a Spin-1/2 degree of freedom with the usual spin operators \(S^x, S^y, S^z\).
The problem geometry, in terms of lattice type, size and boundary conditions.
The Hamiltonian itself. Here, it is naturally expressed as a sum of couplings.
In the following, we guide you towards defining your own custom model, with the above case as an example.
We follow the most direct route, using the CouplingMPOModel framework,
for more flexible alternatives see Details on the implementation of Models.
Note
This Hamiltonian is already implemented as one of the pre-defined models.
Here, we implement it from scratch as an example, but if you wanted to simulate this particular
Hamiltonian in practice, you would use the SpinModel, which has a
more general Hamiltonian and contains our example as a special case.
The first step is to identify what the parameters of your model are.
In this case, we have the coupling constants \(J_x, J_z, h_x, h_z\), and parameters that specify
the lattice geometry (discussed later). In the TeNPy ecosystem, these parameters are
gathered into dictionary-like Config objects, and for the rest of this
guide you can think of model_params as a dictionary of these parameters, e.g.
model_params = {'Jx': 0.5, 'Jz': 1}.
It is common practice to make all parameters optional, in which case you should think about
(and ideally document) the default values for the parameters.
We start implementing our custom model by defining a class for it:
class MyModel(CouplingMPOModel):
r"""The anisotropic spin-1/2 Heisenberg model in an external field.
This is a pedagogical example, and you should probably use the SpinModel instead.
The Hamiltonian is:
.. math ::
H = J_x \sum_i (S^x_i S^x_{i+1} + S^y_i S^y_{i+1})
+ J_z \sum_i S^z_i S^z_{i+1}
- h_x \sum_i S^z_i - h_z \sum_i S^z_i
"""
pass # content will be added later
Note that we define our model as a subclass of CouplingMPOModel.
This means our model inherits all the machinery to build Hamiltonians etc, and we only need
to implement the code that is specific to our model.
The local Hilbert space
The local Hilbert space is represented by a Site (read its doc-string!).
A site defines the meaning of each basis state (i.e. by fixing an order, to define e.g.
that the state are spin_down, spin_up). Additionally, it stores common local operators, such as
\(S^z\) and makes them accessible by name.
We need to tell our model, what its local Hilbert space is.
This is done by implementing the init_sites() method.
It needs to take the model_params as input and returns one Site
instance per site in the unit cell of the lattice (see lattice section below, here this is one site).
The most common sites – e.g. for spins, spin-less or spin-full fermions, or bosons – are predefined
in the module tenpy.networks.site, and in this example we can use one of them directly:
class MyModel(CouplingMPOModel):
def init_sites(self, model_params):
# simple version: no charge conservation
return SpinHalfSite(conserve='None')
If necessary, you can easily extend a pre-defined site by adding further local operators or
completely write your own subclasses of Site.
If you want to use charge conservation (and you probably should, if possible), we need to specify
what charges are conserved at this point already, i.e. we should give a value to the conserve
argument of the site.
Note
If you don’t know about Charge conservation with np_conserved yet, but want to get started with models right away,
you can set conserve=None in the existing sites as above and skip the rest of this
section. If you need a custom site, you can use
leg = tenpy.linalg.np_conserved.LegCharge.from_trivial(d) for an implementation of your
site, where d is the dimension of the local Hilbert space.
In many cases, the possible symmetries we may exploit depend on the
values of the parameters, which is why they are an input to init_sites.
In our example, we can conserve the total \(S^z\) if \(h_x = 0\):
class MyModel(CouplingMPOModel):
def init_sites(self, model_params):
conserve = model_params.get('conserve', 'best')
if conserve == 'best':
if model_params.get('hx', 0) == 0:
conserve = 'Sz'
else:
conserve = None
return SpinHalfSite(conserve=conserve)
Note that we added conserve as a model parameter, such that we can later turn charge
conservation on or off. The possible values for conserve are documented in the site class,
here SpinHalfSite, and it is common to support 'best'
as a value for the conserve model parameter and translate it to the largest possible symmetry,
given the values of the coupling strengths.
Note
The LegCharge of all involved sites need to have a common
ChargeInfo in order to allow the contraction of tensors
acting on the various sites.
This can be ensured with the function set_common_charges().
An example where set_common_charges() is needed would be a coupling
of different types of sites, e.g., when a tight binding chain of fermions is coupled to some
local spin degrees of freedom. Another use case of this function would be a model with a U(1)
symmetry involving only half the sites, say \(\sum_{i=0}^{L/2} n_{2i}\).
The geometry (lattice)
The geometry is usually given by some kind of lattice structure that determines how the sites
are arranged spatially. This implicitly defines e.g. the meaning of a sum over nearest neighbors
\(\sum_{<i, j>}\).
In TeNPy, this is specified by a Lattice class, which contains a unit cell of
a few Sites which are repeated periodically according to the lattice
basis vectors, to form a regular lattice.
Again, we have pre-defined some basic lattices like a Chain,
two chains coupled as a Ladder or 2D lattices like the
Square, Honeycomb and
Kagome lattices; but you are also free to define your own generalizations.
See Details on the lattice geometry.
By default, the CouplingMPOModel puts your model on
a Chain, and looks for its length as model_params['L'].
If you want to use a different pre-defined lattice, you can put it into the parameters, e.g.
as model_params['lattice'] = 'Square', and the size is taken from model_params['Lx']
and model_params['Ly'], while the boundary conditions are model_params['bc_x']
and model_params['bc_y'].
Of course, simply changing the lattice only makes sense if the Hamiltonian is defined in a lattice
independent language, e.g. in terms of “nearest neighbor pairs”.
As we will explore in the next section, this is in fact the natural way to define Hamiltonians in TeNPy.
It is also common to have specialized classes for special lattices:
class MyModelKagome(MyModel):
default_lattice = Kagome
force_default_lattice = True
def init_sites(self, model_params):
# note: Kagome has three sites per unit-cell
site = MyModel.init_site(model_params)
return (site, site, site)
Setting default_lattice = Kagome means that the lattice defaults to Kagome, if 'lattice' not in model_params,
while setting force_default_lattice = True means that this model does not allow any other
lattice. Thus, MyModelKagome does what its name promises to do.
For custom lattices, or more complicated code, you can overwrite the
init_lattice() method, similar to how we did
for init_sites above.
The Hamiltonian
The last ingredient we need to implement for a custom model is its Hamiltonian.
To that end, we override the init_terms() method.
At this point during model initialization, the lattice is already initialized, and we
may access self.lat and use e.g. the pairs attribute
for convenient definition of couplings between e.g. nearest-neighbor pairs.
There are a bunch of convenience methods implemented in CouplingModel,
which make this easy. Let us summarize them here:
add_onsite()for a sum of onsite terms \(\sum_i h_i \hat{A}_i\).add_coupling()for a sum of two-body couplings \(\sum_i J_i \hat{A}_i \hat{B}_{i+n}\).add_multi_coupling()for a sum of multi-body couplings \(\sum_i J_i \hat{A}_i \hat{B}_{i+n} ... \hat{F}_{i+m}\).
Note
A single call to each of these methods adds an extensive number of terms to your Hamiltonian,
as it includes a sum over all sites in the definition.
This means that a Hamiltonian like \(H = -3 \sum_i S_i^z\) is realized as a single call to
add_onsite(), without an explicit loop over i.
Note
These methods allow the prefactors to be site-dependent; you can either give a single number
as the prefactor, or a list/array that is tiled to fit the size.
E.g. if an onsite term with strength=1 gives you a uniform magnetic field,
strength=[1, -1] gives you the corresponding staggered field,
assuming a chain of even length.
add_local_term()for a single term \(\hat{A}_i\) or \(\hat{A}_i \hat{B}_{j}\) or \(\hat{A}_i \hat{B}_{j} ... \hat{F}_k\).
Warning
You probably should not directly use add_onsite_term(),
add_coupling_term() and
add_multi_coupling_term().
They do not handle Jordan-Wigner strings and they need MPS indices as inputs, not
lattice positions.
See also add_exponentially_decaying_coupling()
Note
Instead of a single operator name like 'Sx', you can put multiple operator names
separated by whitespace to represent the product of these operators.
For example, self.add_onsite(-2.j * (-hz), u, 'Sx Sy') is equivalent to (but worse than)
to self.add_onsite(-hz, u, 'Sz').
For our example, we define the Hamiltonian by implementing:
class MyModel(CouplingMPOModel):
def init_sites(self, model_params):
...
def init_terms(self, model_params):
Jx = model_params.get('Jx', 1.)
Jz = model_params.get('Jz', 1.)
hx = model_params.get('hx', 0.)
hz = model_params.get('hz', 0.)
for u in range(len(self.lat.unit_cell)):
self.add_onsite(-hx, u, 'Sx')
self.add_onsite(-hz, u, 'Sz')
for u1, u2, dx in self.lat.pairs['nearest_neighbors']:
self.add_coupling(Jz, u1, 'Sz', u2, 'Sz', dx)
# Sx and Sy violate Sz conservation.
# need to define them using Sp = Sx + i Sy, Sm = Sx - i Sy
# Sx.Sx + Sy.Sy = .5 * (Sp.Sm + Sm.Sp) = .5 * (Sp.Sm + hc)
self.add_coupling(.5 * Jx, u1, 'Sp', u2, 'Sm', dx, plus_hc=True)
Note
If we did not care about charge conservation, we could have also done
add_coupling(Jx, u1, 'Sx', u2, 'Sx', dx) and
add_coupling(Jx, u1, 'Sy', u2, 'Sy', dx).
This only works if we set conserve='None' or conserve='parity',
as otherwise the site does not even define 'Sx'.
Also, note that that the on-site operators Sp=\(S^+_i\) and Sm=\(S^-_i\)
do not conserve the total \(S^z\), but you can still use them to define the combined
coupling \(S^+_i S^-_j\) that does conserve \(S^z\).
At this point we are done defining our model, and have reproduced the result at the very top of the chapter. We should, however, make sure that we defined the model correctly.
Verifying models
Especially when you define custom models, we strongly recommend you triple-check if you correctly implemented the model you are interested in (i.e. have the correct couplings between correct sites). This is a crucial step to make sure you are in fact simulating the model that you are thinking about and not some random other model with entirely different physics.
Note
If the model contains Fermions, you should read the introduction to Fermions and the Jordan-Wigner transformation.
To verify that you have added the correct terms, initialize the model on a small lattice (we also set \(J_x=0\) here for readability, but you should turn it on to verify the full model), e.g.:
model = MyModel({'lattice': 'Square', 'Lx': 2, 'Ly': 3, 'Jx': 0, 'hz': 0.2})
Now, print all couplings and onsite terms in the model to console:
print(model.all_coupling_terms().to_TermList() + model.all_onsite_terms().to_TermList())
Which gives you the following output for our example:
1.00000 * Sz_0 Sz_1 +
1.00000 * Sz_0 Sz_2 +
1.00000 * Sz_0 Sz_3 +
1.00000 * Sz_1 Sz_2 +
1.00000 * Sz_1 Sz_4 +
1.00000 * Sz_2 Sz_5 +
1.00000 * Sz_3 Sz_4 +
1.00000 * Sz_3 Sz_5 +
1.00000 * Sz_4 Sz_5 +
-0.20000 * Sz_0 +
-0.20000 * Sz_1 +
-0.20000 * Sz_2 +
-0.20000 * Sz_3 +
-0.20000 * Sz_4 +
-0.20000 * Sz_5
You may be surprised to see nine different two-body couplings on this 2 x 3 square patch.
Let us look at the couplings in detail to figure out why this might be.
We need to understand the meaning of the site indices, i.e. where does Sz_4 live spatially?
The convention for site indices comes from the MPS geometry and may be hard to read.
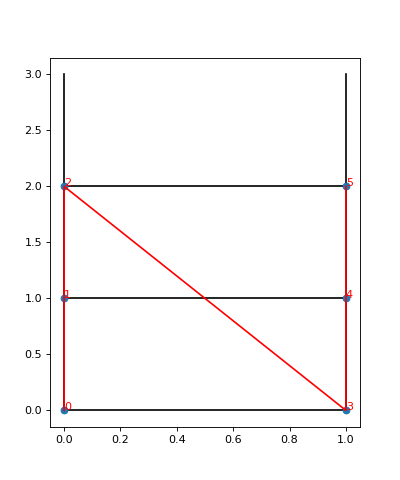
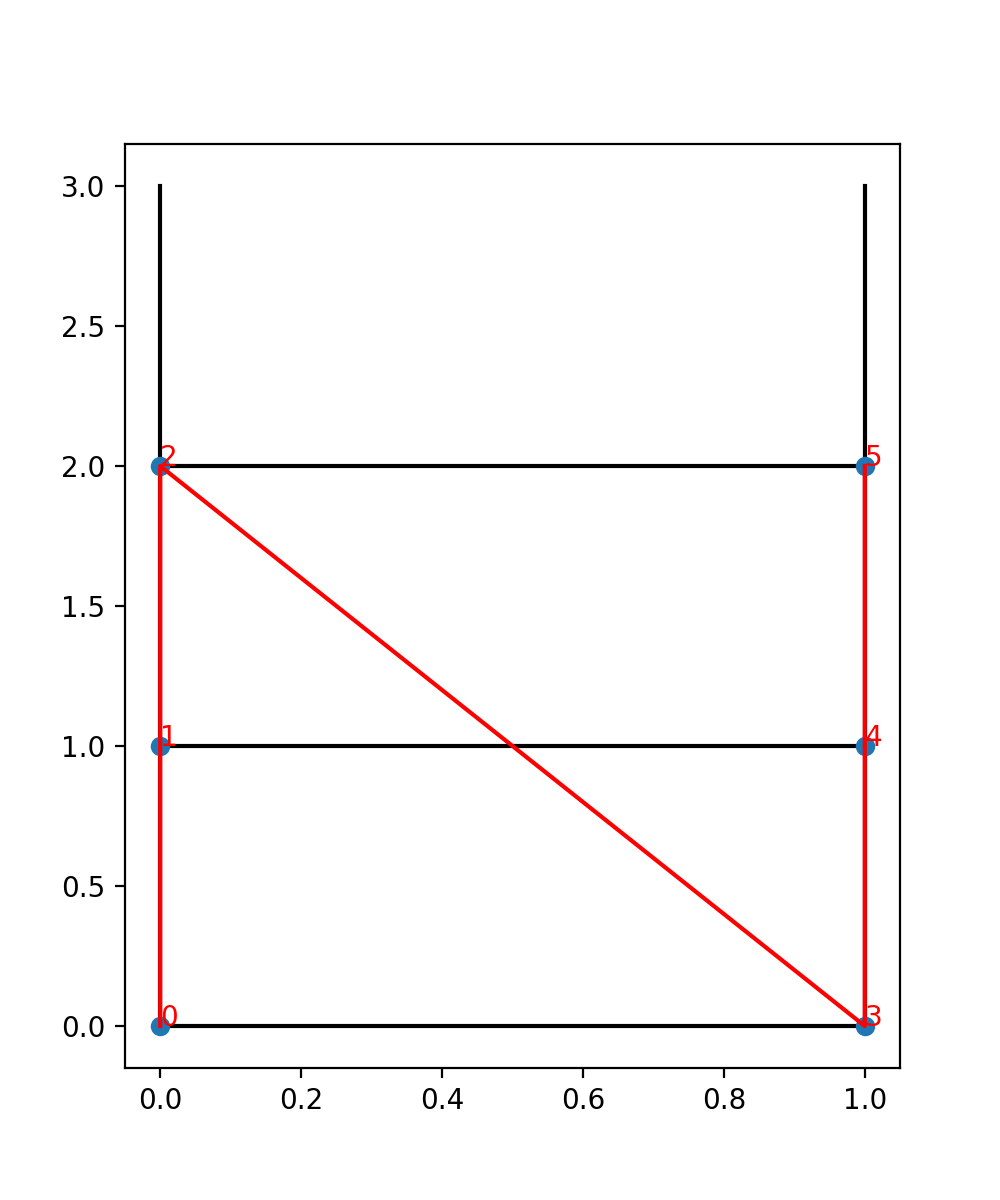
To visualize the site order of the lattice, you may run the following snippet:
import matplotlib.pyplot as plt
plt.figure(figsize=(5, 6))
ax = plt.gca()
model.lat.plot_coupling(ax)
model.lat.plot_sites(ax)
model.lat.plot_order(ax)
plt.show()
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
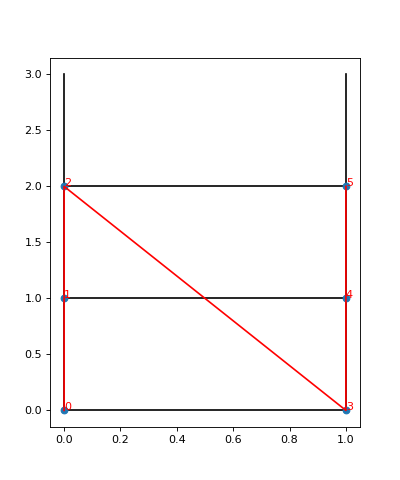
We see the lattice plotted in black. Concretely, we get a black line for each pair of nearest-neighbor sites. The red line goes through the sites in order, and we see the site indices labelled.
In particular, we can now understand why we get nine different couplings; we see from the plot
that the lattice has open boundaries in x-direction but periodic boundaries in y-direction.
Try playing around with different boundary conditions, e.g.
MyModel({'lattice': 'Square', 'Lx': 2, 'Ly': 3, 'Jx': 0, 'bc_y': 'open'})
or
MyModel({'lattice': 'Square', 'Lx': 2, 'Ly': 3, 'Jx': 0, 'bc_x': 'periodic'}).
See Details on the lattice geometry regarding boundary conditions.
You can also use get_numpy_Hamiltonian() to see if the Hamiltonian
is what you expect it to be.
You will need to choose a relatively small system for the full Hamiltonian to fit into RAM.
This is strongly recommended if you defined your own operators, as e.g. the
PXPChain does.
Contribute your model?
If you have implemented a model, and you think it may be useful to the broader community, consider
contributing it to TeNPy via a pull request.
We have Coding Guidelines, and you can have a look at the implementation
of e.g. the SpinModel as a guide, but do not let formalities
stop you from sharing your code, we can always address any nitpicks ourselves.
Further Reading
Details and ideas behind the implementation: Details on the implementation of Models
Look at the implementation of the pre-defined models in
tenpy.models. Most are based on theCouplingMPOModelas discussed here.The
AKLTChainis a notable counter-example where it is actually easier to defineH_bondthan to write down couplings.If the Hamiltonian is already given in MPO form (e.g. because it comes from some other software), it can be used to directly build a model, as is done in
examples/mpo_exponentially_decaying.py.